
鉅大LARGE | 點擊量:4827次 | 2018年08月03日





為何存在電池電壓衰減的情況
研究亮點:
電壓衰減的根源為失氧
失氧造成所有過渡金屬的平均價態不斷變低(Ni3+/Ni4+電對轉化為Ni2+/Ni3+電對,Mn4+/Mn5+電對轉化為Mn3+/Mn4+電對,Co3+/Co4+電對轉化為Co2+/Co3+電對),導致電壓衰減
失氧還會造成材料顆粒的微瑕疵(如在顆粒內部形成大孔),導致電壓衰減
技術亮點:
同步X射線吸收光譜(XAS)具有加大的穿透功率,能穿透整個陰極,提供體相分析
STEM具有原子層面的空間分辨率,提供表層分析(5nm)
新型三維電子斷層成像技術,觀察孔隙在三維空間上的生長
電壓衰減已經成為阻止一系列高能量密度電池電極商業化的重要原因。富鋰材料,得益于較高的容量(>250mAhg-1vs.<200mAhg-1),是替代傳統陰極材料的理想選擇。但其在循環過程中,平均電壓不斷衰減,嚴重影響能量效率且給電池管理系統帶來困難。電壓為何衰減?近日,布魯克爾海文國家實驗室聯合中科院物理所和阿貢國家實驗室,使用X射線光譜和三維電子顯微鏡成像技術,原位和非原位探索材料在充放電過程中,體相和表面的變化。
氧化還原電對的演變
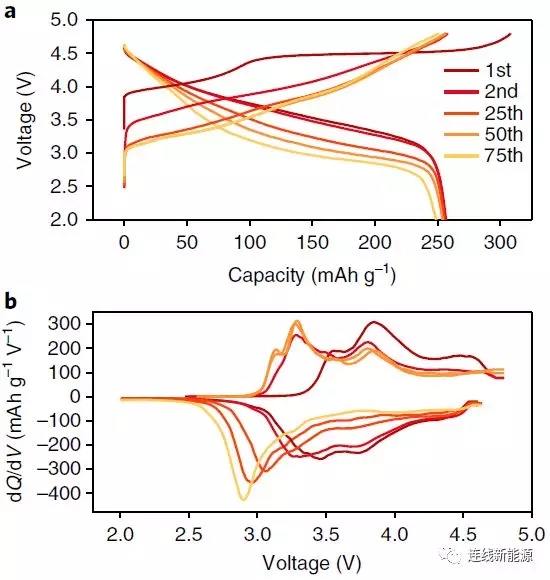
圖1Li1.2Ni0.15Co0.1Mn0.55O2的電化學性:a.充放電曲線;b.dQ/dV圖。電壓衰減問題顯而易見
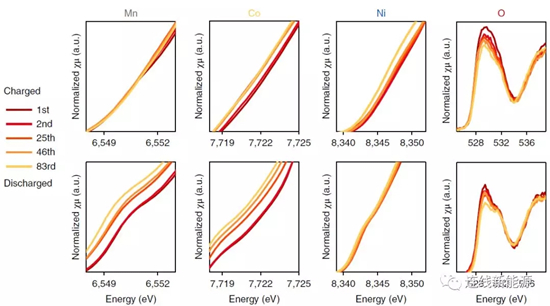
圖2Li1.2Ni0.15Co0.1Mn0.55O2中不同元素的XAS結果。隨循環進行,三種金屬的平均價態持續降低,證明體相中過渡金屬和氧的配位能力在減弱。
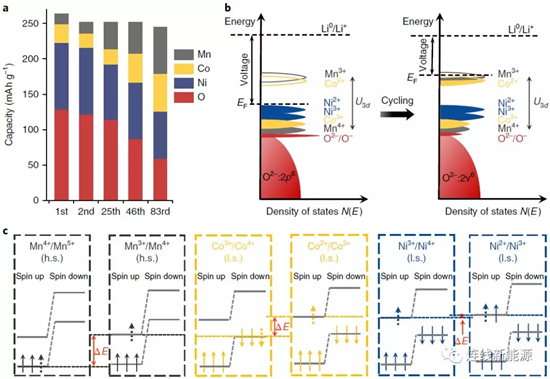
圖3Li1.2Ni0.15Co0.1Mn0.55O2在循環過充中,氧化還原電對的演化:a.每種元素對放電容量的貢獻;b.費米能級由于電子結構的變化而升高;c.不同氧化還原電對得失電子時涉及到的不同能級
如圖3a,首周時,O和Ni對容量貢獻占主導,然而,隨循環進行,Mn和Co對容量貢獻增多,逐漸補償O和Ni對容量貢獻的減少,最后Co和Mn的容量占主導地位。氧化還原電對從O和Ni逐漸轉變成Mn和Co,勢必會影響電壓分布。如圖3b,陰極材料的能態密度在不斷改變。電池的開路電壓(OCV)由費米能級相對于Li+/Li0能級的差別決定,與將電子從富鋰陰極移動到Li陽極的功焓有關。起初,材料的費米能級位于Ni2+/Ni3+氧化還原電對之上,循環后,發生氧氣釋放并導致TM還原。如Ni傾向于首先在表面還原,形成電子絕緣的非活性巖鹽相,降低Ni對容量的貢獻;Mn和Co的還原產生Mn3+/Mn4+和Co2+/Co3+氧化還原電對,這種還原能將費米能級變得更高,導致OCV變低以及工作電壓變低。這個過程也能解釋為什么O對容量的貢獻逐漸減少,由于TM還原,TM與O的共價鍵減弱,造成參與氧化還原的O減少。
比較圖3c不難發現,Ni2+/Ni3+與Ni3+/Ni4+電對的能級差最小(△E),而Co2+/Co3+和Co3+/Co4+電對的能級差加大,Mn3+/Mn4+與Mn4+/Mn5+電對的能級差更大。能級差的不同是因為電子結構不同。對Mn和Co而言,從一個電對變到另一個,需要設計不同的軌道。如Co2+/Co3+電對變成Co3+/Co4+電對,涉及到在自旋向上eg軌道上失去(氧化)或得到(還原)一個電子。然而對于Ni,Ni2+/Ni3+電對與Ni3+/Ni4+電對的轉變,涉及的電子得失是在同一軌道(自旋向上eg)。因此,Mn和Co是OCV下降和所謂電壓衰減主要原因。
表面反應
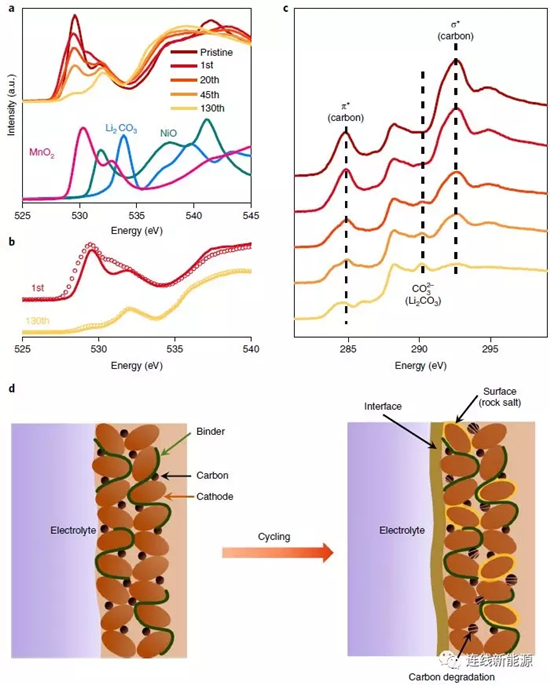
圖4Li1.2Ni0.15Co0.1Mn0.55O2表層分解導致過電位增大:a.富鋰材料在充電態下的O譜及各參考組分的O譜;b.充電態(圈)與放電態(線)的O譜;c.C譜;d.循環前后的示意圖,循環后形成新界面、表面發生重組且導電碳降解
前面所述的XAS能提供豐富的體相反應信息,而軟XAS技術則能對材料的表面進行表征(5nm)。O譜所有的峰如圖4a所示,隨循環進行,代表MnO2的峰不斷減弱,而代表NiO的特征峰不斷增強,證明表面結構發生重組,巖鹽結構因此生成。圖4b中代表Li2CO3的峰(535eV以上)逐漸變強,代表表面OH物種增多,是因為電解液分解而形成不同的有機/無機化合物,如Li2CO3、RCO2Li等。C譜所有的峰如圖4c所示,284.8eV和292.6eV歸結于導電碳的π反成鍵和σ反成鍵,290.2eV歸結于Li2CO3中的CO32-。隨循環進行,導電碳的特征峰減弱,證明導電碳黑逐漸分解,可能是因為在高電壓下,PF6-嵌入石墨中。相反,歸屬于Li2CO3的峰增強,Li2CO3代表陰極界面膜(CEI)的產物,意味著CEI隨循環進行在不斷生長。CEI的增長會使電化學反應動力學更加滯后,造成過電勢增加且電壓衰減。
微結構瓦解的開始和蔓延
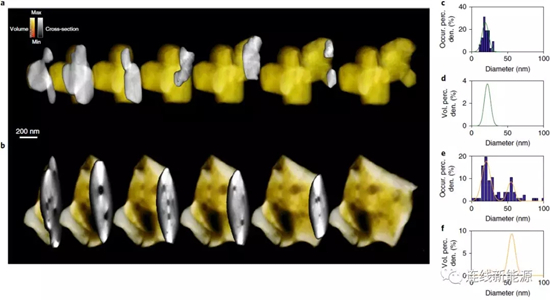
圖5Li1.2Ni0.15Co0.1Mn0.55O2材料的三維斷層圖:a.初始態和b.循環15周后的顆粒截面圖;內部孔徑分布圖(c、d)初始態和(e、f)循環15周后
失氧怎樣誘使材料的微觀結構發生改變?研究人員采用ADF-STEM成像和空間分辨EELS來研究結構的成核和演變過程。初始態和循環15周后,陰極材料的3D內部結構如圖5a、b所示,圖5b能觀察到循環后顆粒內部產生一些大孔。通過定量分析孔徑,可推斷這些大孔在一開始并不存在(圖5c、d),但15周時,大孔占據材料孔隙率的絕大部分(圖5e、f)。這種大孔是在循環的過程中產生的,很可能是由失氧所留下的成核空位演化而成的。
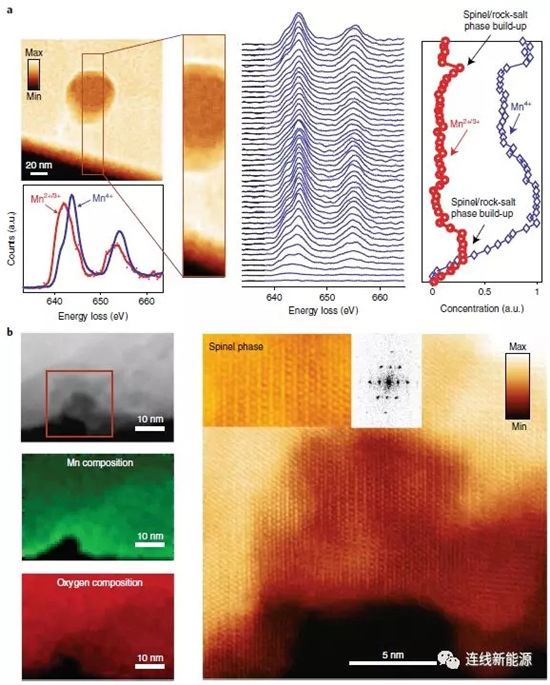
圖6顆粒表面暴露的孔隙(a)和體相隱藏的孔隙(b)的空間分辨EELS分布
為進一步弄清這些大孔的成因,科研人員采用STEM-EELS技術來分析材料顆粒中的孔結構,例如,研究對象為顆粒體相中一個隱藏的孔隙和顆粒表面一個暴露的孔隙。圖6a顯示隱藏的孔周圍形成了一層薄的Mn2+殼。由于這些小孔隙發現于初始材料中,故形成于材料的制備過程,其形狀和大小保持不變直至在其周圍開始產生氧氣(循環過程中)。與之對應的是暴露的孔隙,這些孔隙與電解液接觸,會逐漸形成一層厚的尖晶石相/巖鹽相,因此,EELS相對濃度分布表明,表面體積中的Mn相對含量增加。其實,很多孔隙既不是完全隱藏也不是完全暴露的。這種部分暴露的孔隙周圍,存在氧氣擴散通道(以微結構瑕疵、位錯和晶界的形式存在),隨循環進行,孔會變多且變大,與那些完全暴露的孔隙一起,加速結構的相變、O2的進一步釋放、微結構瑕疵的產生和電壓衰減。
如何抑制失氧
失氧是電壓衰減的根源,怎樣才能抑制失氧?一種方法是對材料表/界面進行處理。例如,表面包覆能減少電解液與界面的直接接觸。注意最好對初級顆粒進行包覆,而非次級顆粒。除包覆外,還可以在顆粒表面引入氧空位,這種表面處理方式能顯著減少失氧。另一種方法就是體相摻雜。例如,摻雜Al陽離子,Al能占據四面體位點,因此能抑制過渡金屬離子向四面體空位遷移,這種遷移過程是伴隨相轉變過程與失氧過程同時發生的。
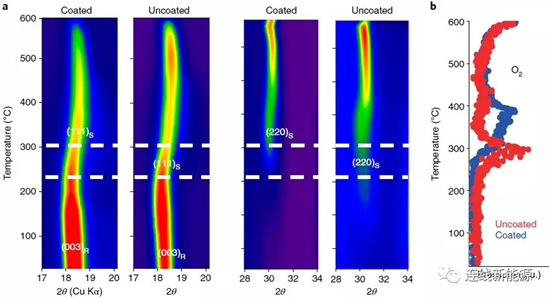
圖7Li1.2Ni0.15Co0.1Mn0.55O2材料與經AlF3包覆后的熱穩定性對比:a.加熱過程中的原位XRD說明沒包覆的材料更快經歷相變(R代表斜方六面體相,S代表尖晶石相);b.加熱過程中的氧釋放情況,沒包覆的材料在更低的溫度下就會釋放氧。
無論測試循環性能測試還是熱穩定性測試,都涉及氧的釋放和隨之引起的過渡金屬的還原、遷移。相比之下,熱穩定性測試的耗時短、用量少,因此可以替代循環測試,評估材料的結構穩定性。材料熱穩定性的高低可直接用來評估電壓衰減的程度。
結論
失氧是引起電壓衰減問題的根源。氧釋放會造成材料更高的比表面以及使顆粒內部產生更多的缺陷,這將進一步加速氧釋放過程;氧釋放引起Mn和Co的還原(向低價態電對Mn3+/Mn4+和Co2+/Co3+轉化),造成電壓衰減;此外,氧釋放加速表面重組,陰極界面膜(CEI)的傳質變慢且電化學性能變差,造成電壓衰減。今后的科研工作應圍繞抑制氧釋放展開,主要途徑為表/界面處理或體相摻雜。
上一篇:讓快充更加安全的新型鋰電池材料
下一篇:動力電池業兩大新規今起實施


















